Seqera AI
Austyn Trull, Lara Ianov, Ph.D.
Introduction
Discussion Question
How many of you have used AI to aid you in learning bioinformatics? What are some pros and cons?
Seqera AI is a bioinformatics-focused assistant designed for all stages of Nextflow development. Unlike general AI coding tools, it’s tightly integrated with Nextflow best practices and nf-core standards. It can suggest validated pipelines, generate DSL2-compliant Nextflow code, execute tests, help users troubleshoot error messages and much more.
Currently Seqera AI can be used in two ways: 1. Through the online platform, https://seqera.io/ask-ai/chat (note that the free version is limited to 100 chats per month) 2. Through the Nextflow VS Code extension.
Here is a brief summary of some common use cases for each AI platform:
Seqera AI online
- Full-featured web environment for pipeline exploration and initial development (including connection to pipelines hosted in GitHub)
- Ability to test code snippets and validate pipeline components
- SRA dataset search with natural language queries
Nextflow VS Code Extension
- Provides IDE-native experience which can facilitate in-depth pipeline development
- Real-time support for coding
- Direct access to log files and terminal outputs can enhance debugging
A more in-depth overview can be found at the following links:
- https://seqera.io/blog/legacy-scripts-to-nextflow-seqera-ai/
- https://seqera.io/blog/seqera-ai--nextflow-vs-code/
Something to take note of is that the behavior of each platform may slightly differ. We find that the online platform is great for more in-depth answers, learning, exploration and understanding concepts, while the VS Code extension is better for context-specific support during active development. Therefore, they complement each other.
For the purposes of this workshops, the example below used the Seqera AI online.
Writting a custom configuration file
A key strength of Nextflow, is its high level of modularity. While an in-depth overview of this topic is beyond the scope of this workshop, a common use-case is the ability of over-writing pipeline defaults with a custom configuration file.
An example of a custom configuration file which has already been used
in our example run is the cheaha profile. The key
distinction between an institutional profile and a custom configuration
file lies in how they are managed and accessed. Institutional profiles
are centrally hosted and maintained, making them easily accessible as
shared resources. They are enabled using the -profile flag.
In contrast, custom configuration files are local to the user’s
environment and must be explicitly provided with the -c
flag. Essentially, institutional profiles simplify access and ensure
consistency across users, while custom configs offer flexibility for
local customization.
With that in mind, some common use cases where a custom configuration file is needed includes request for increased computational resources and/or custom parameters for the underlying tools of the pipeline.
As an example, let’s assume that we would like to increase the
threads and decrease the memory requirements for the STAR.
How do you do this? Seqera AI can help:
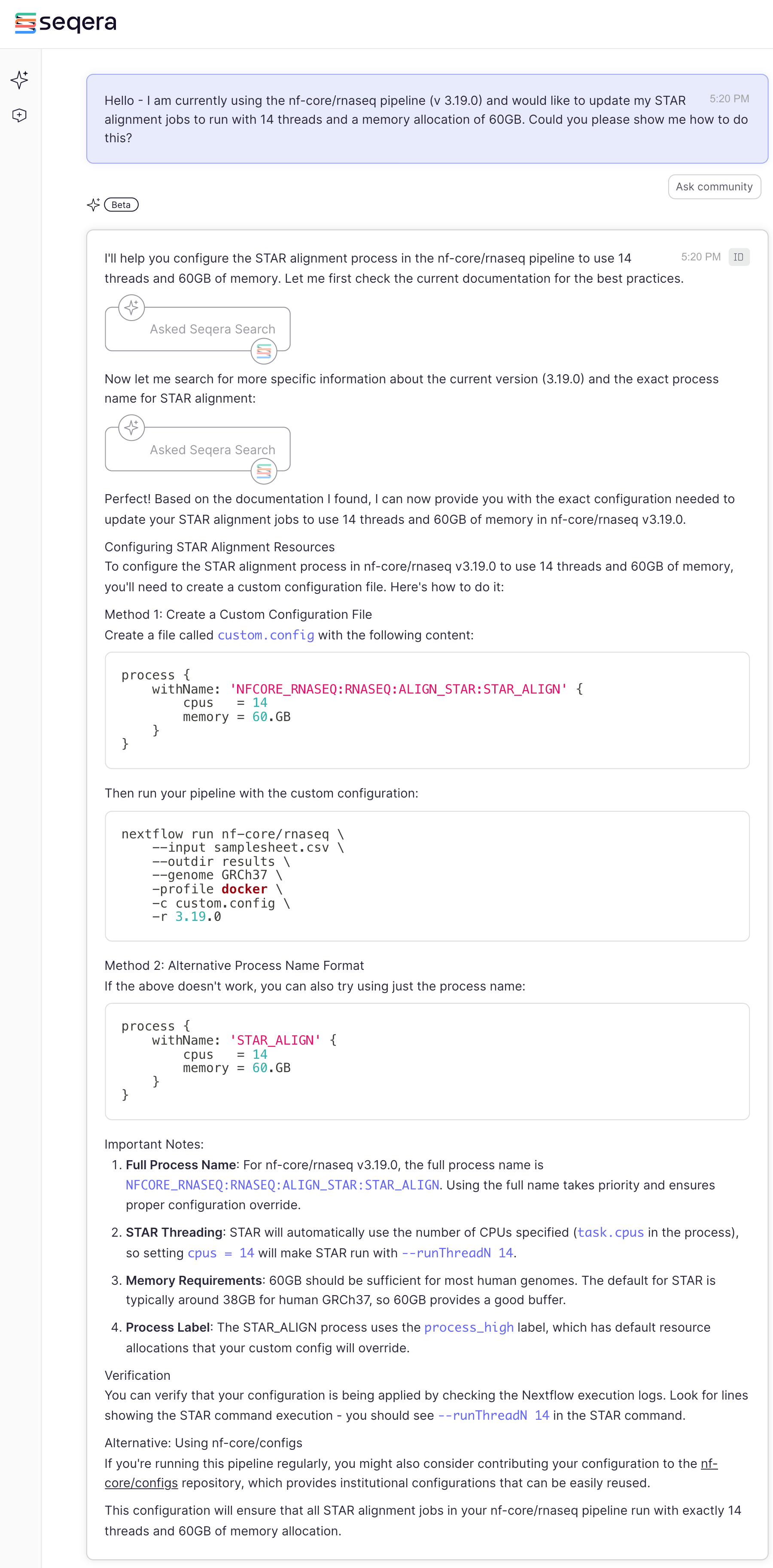
Note that we have specified the version of the pipeline which was used in the example run for this workshop. This specific context can aid the AI in providing more refined answers.
The answer provided a detailed description, and an alternative
approach should method 1 not work. It also provided a sample run
command, but to adapt the answer to our run_rnaseq.sh, you
would need to simply include the -c custom.config to our
existing script:
#!/usr/bin/env bash
#SBATCH --job-name=nfcore_rnaseq
#SBATCH --output=nfcore_rnaseq.out
#SBATCH --error=nfcore_rnaseq.err
#SBATCH --time=12:00:00
#SBATCH --partition=short
#SBATCH --mem-per-cpu=5000
#SBATCH --cpus-per-task=1
# load environment
module load Anaconda3
conda activate $USER_SCRATCH/conda_envs/nfcore_workshop
# run workflow
nextflow run nf-core/rnaseq \
--outdir ./results \
-profile cheaha \
-r 3.19.0 \
-params-file ./params.yml \
-c custom.config ## custom configImportantly it also provided a great tip - to verify
that your modified configuration is actually being applied. Indeed the
best approach to do this is check the STAR command in this
case. With that in mind let’s move forward to the next example where AI
can also be helpful.
Navigation of the work dir
The example above touched on a custom configuration for computational resources. What about tool parameters? The answer is the same as above - you can use the same syntax in a custom configuration file to add tool-specific parameters.
Notably, nf-core/rnaseq is nf-core’s most matured pipeline. As such, the developers have already made adding tool-specific parameters very straight-forward and we have already implemented them in our test run by including the following:
Importantly, this did not become an option until versions >= 3.10.
So what would you do if the pipeline did not have these
extra params (which is very common across many other
nf-core pipelines)? Once again, the custom.config file
would be the solution, e.g.:
process {
// Salmon post STAR alignment
withName: 'NFCORE_RNASEQ:RNASEQ:QUANTIFY_STAR_SALMON:SALMON_QUANT' {
ext.args = '--gcBias --seqBias'
}
// Salmon in quasi-mapping mode
withName: 'NFCORE_RNASEQ:RNASEQ:QUANTIFY_PSEUDO_ALIGNMENT:SALMON_QUANT' {
ext.args = '--gcBias --seqBias'
}
}After the run is completed, how can we be certain that this
configuration was successful? While there are a couple of locations
which you can find the command for a process, we find that checking the
work directory is best since it also contains tool-specific
logs along with Nextflow’s .command.sh file. Navigating
this directory can be also very useful for troubleshooting or output
exploration.
Given the complexity of the work directory, let’s ask
Seqera AI to help us parse through the data:
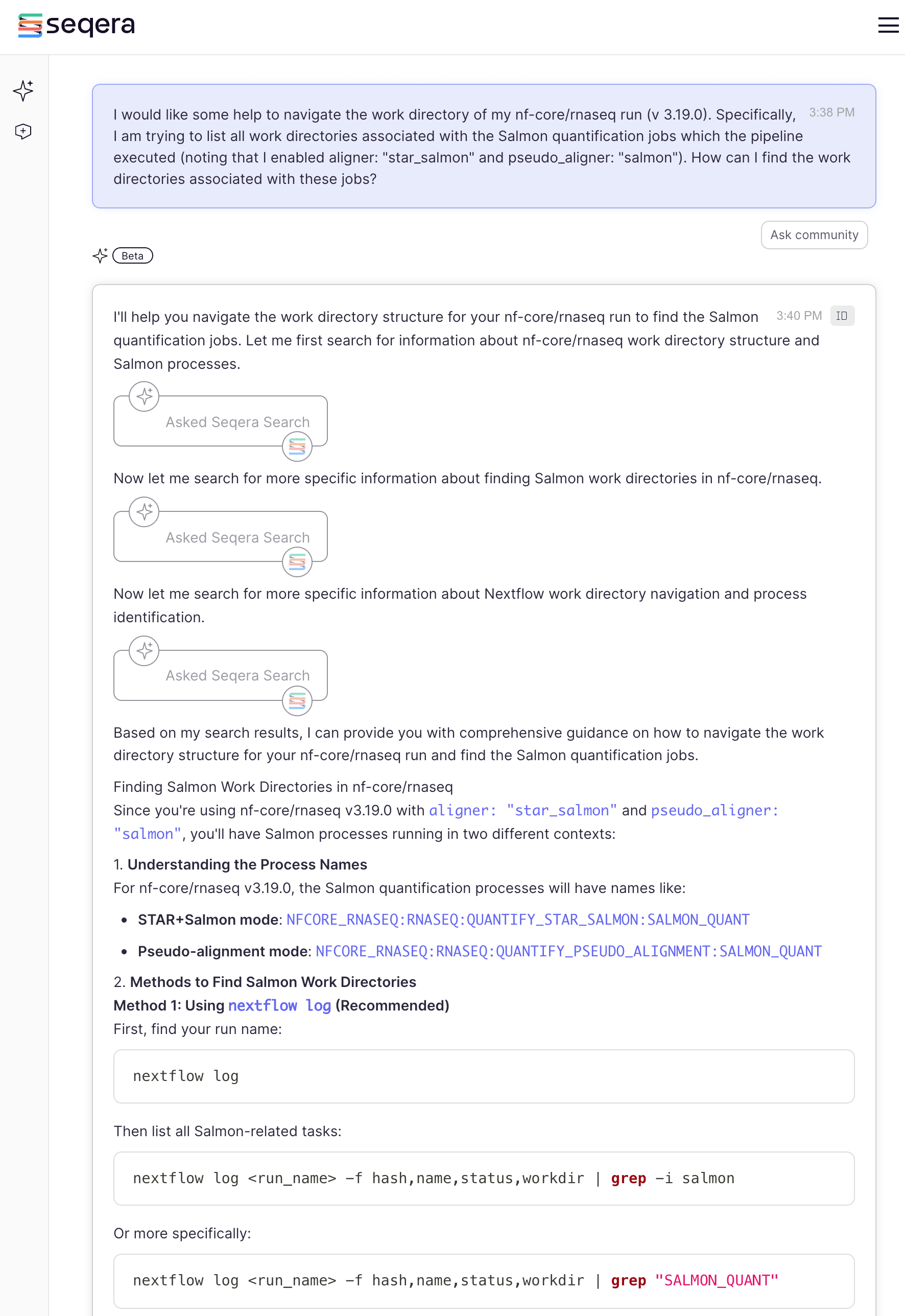
The answer provides multiple methods, but method #1 shown above is the best suggestion. Let’s try the suggestion from our example run:
TIMESTAMP DURATION RUN NAME STATUS REVISION ID SESSION ID COMMAND
2025-07-24 16:57:32 2h 36m 3s irreverent_lovelace OK 0bb032c1e3 db1ce7d9-5e40-4205-98cc-8d06ceed6183 nextflow run nf-core/rnaseq --outdir ./results -profile cheaha -r 3.19.0 -params-file ./params.ymlThe output displays all SALMON_QUANT work
directories. The ones which we would want to explore are linked to the
aligment or quasi-mapping jobs (QUANTIFY_STAR_SALMON,
QUANTIFY_PSEUDO_ALIGNMENT):
e4/77fd58 NFCORE_RNASEQ:RNASEQ:FASTQ_QC_TRIM_FILTER_SETSTRANDEDNESS:FASTQ_SUBSAMPLE_FQ_SALMON:SALMON_QUANT (N01_AM_Naive) COMPLETED /scratch/lianov/nfcore_rnaseq/example_run/work/e4/77fd581558c8d74aa9574f4660d815
8a/c60514 NFCORE_RNASEQ:RNASEQ:FASTQ_QC_TRIM_FILTER_SETSTRANDEDNESS:FASTQ_SUBSAMPLE_FQ_SALMON:SALMON_QUANT (N04_AM_Naive) COMPLETED /scratch/lianov/nfcore_rnaseq/example_run/work/8a/c605145ac7afdd4e329d8f3732c1c1
23/591642 NFCORE_RNASEQ:RNASEQ:FASTQ_QC_TRIM_FILTER_SETSTRANDEDNESS:FASTQ_SUBSAMPLE_FQ_SALMON:SALMON_QUANT (R07_AM_Allo24h) COMPLETED /scratch/lianov/nfcore_rnaseq/example_run/work/23/591642d9b26fbd9555d290ad7b99df
a6/b0920b NFCORE_RNASEQ:RNASEQ:FASTQ_QC_TRIM_FILTER_SETSTRANDEDNESS:FASTQ_SUBSAMPLE_FQ_SALMON:SALMON_QUANT (N02_AM_Naive) COMPLETED /scratch/lianov/nfcore_rnaseq/example_run/work/a6/b0920b19805b7b7bd5550668e81237
72/7691b6 NFCORE_RNASEQ:RNASEQ:FASTQ_QC_TRIM_FILTER_SETSTRANDEDNESS:FASTQ_SUBSAMPLE_FQ_SALMON:SALMON_QUANT (N03_AM_Naive) COMPLETED /scratch/lianov/nfcore_rnaseq/example_run/work/72/7691b695a7c26eb55e7c1474615255
2f/cedf73 NFCORE_RNASEQ:RNASEQ:FASTQ_QC_TRIM_FILTER_SETSTRANDEDNESS:FASTQ_SUBSAMPLE_FQ_SALMON:SALMON_QUANT (R08_AM_Allo24h) COMPLETED /scratch/lianov/nfcore_rnaseq/example_run/work/2f/cedf7322607bb5ba42497733fd8171
cd/783419 NFCORE_RNASEQ:RNASEQ:FASTQ_QC_TRIM_FILTER_SETSTRANDEDNESS:FASTQ_SUBSAMPLE_FQ_SALMON:SALMON_QUANT (R06_AM_Allo24h) COMPLETED /scratch/lianov/nfcore_rnaseq/example_run/work/cd/7834197d28fa8b1fe51ac9657bd9fd
8e/7c2a24 NFCORE_RNASEQ:RNASEQ:FASTQ_QC_TRIM_FILTER_SETSTRANDEDNESS:FASTQ_SUBSAMPLE_FQ_SALMON:SALMON_QUANT (R05_AM_Allo24h) COMPLETED /scratch/lianov/nfcore_rnaseq/example_run/work/8e/7c2a2475d31255e520892cbab6d929
5c/360f85 NFCORE_RNASEQ:RNASEQ:QUANTIFY_PSEUDO_ALIGNMENT:SALMON_QUANT (N01_AM_Naive) COMPLETED /scratch/lianov/nfcore_rnaseq/example_run/work/5c/360f85ae4c26c50f6a7f9d20729eda
98/b0c9bd NFCORE_RNASEQ:RNASEQ:QUANTIFY_PSEUDO_ALIGNMENT:SALMON_QUANT (R07_AM_Allo24h) COMPLETED /scratch/lianov/nfcore_rnaseq/example_run/work/98/b0c9bde47b876e6c3b9d499b045f94
72/bacb86 NFCORE_RNASEQ:RNASEQ:QUANTIFY_PSEUDO_ALIGNMENT:SALMON_QUANT (N04_AM_Naive) COMPLETED /scratch/lianov/nfcore_rnaseq/example_run/work/72/bacb86faec759040a45f044739ed4e
f1/56043a NFCORE_RNASEQ:RNASEQ:QUANTIFY_PSEUDO_ALIGNMENT:SALMON_QUANT (N02_AM_Naive) COMPLETED /scratch/lianov/nfcore_rnaseq/example_run/work/f1/56043ae4f7c9f2f30e636c365e39d2
ea/9ca6cc NFCORE_RNASEQ:RNASEQ:QUANTIFY_PSEUDO_ALIGNMENT:SALMON_QUANT (N03_AM_Naive) COMPLETED /scratch/lianov/nfcore_rnaseq/example_run/work/ea/9ca6ccb8ff8296337f95792dc74380
50/ecada6 NFCORE_RNASEQ:RNASEQ:QUANTIFY_PSEUDO_ALIGNMENT:SALMON_QUANT (R06_AM_Allo24h) COMPLETED /scratch/lianov/nfcore_rnaseq/example_run/work/50/ecada623996e43d1d842f5266a6547
51/3cab4e NFCORE_RNASEQ:RNASEQ:QUANTIFY_PSEUDO_ALIGNMENT:SALMON_QUANT (R05_AM_Allo24h) COMPLETED /scratch/lianov/nfcore_rnaseq/example_run/work/51/3cab4eb44b8e63a563053501f437f6
fa/b98b9e NFCORE_RNASEQ:RNASEQ:QUANTIFY_PSEUDO_ALIGNMENT:SALMON_QUANT (R08_AM_Allo24h) COMPLETED /scratch/lianov/nfcore_rnaseq/example_run/work/fa/b98b9eea35cde7ac6b7112c6773490
57/8fc99c NFCORE_RNASEQ:RNASEQ:QUANTIFY_STAR_SALMON:SALMON_QUANT (R07_AM_Allo24h) COMPLETED /scratch/lianov/nfcore_rnaseq/example_run/work/57/8fc99c41b020e616eaad102f5b97ba
b9/ec86d6 NFCORE_RNASEQ:RNASEQ:QUANTIFY_STAR_SALMON:SALMON_QUANT (N01_AM_Naive) COMPLETED /scratch/lianov/nfcore_rnaseq/example_run/work/b9/ec86d64896b1dc36f35ab5ffd9b558
06/0fed5c NFCORE_RNASEQ:RNASEQ:QUANTIFY_STAR_SALMON:SALMON_QUANT (N03_AM_Naive) COMPLETED /scratch/lianov/nfcore_rnaseq/example_run/work/06/0fed5c11b435ef09e2e1691dacd403
6e/17b9b8 NFCORE_RNASEQ:RNASEQ:QUANTIFY_STAR_SALMON:SALMON_QUANT (N04_AM_Naive) COMPLETED /scratch/lianov/nfcore_rnaseq/example_run/work/6e/17b9b8f291a9ef7a4469dee5ed5c15
b4/619e8f NFCORE_RNASEQ:RNASEQ:QUANTIFY_STAR_SALMON:SALMON_QUANT (N02_AM_Naive) COMPLETED /scratch/lianov/nfcore_rnaseq/example_run/work/b4/619e8fafdf1618e723df8981533a24
e7/d39252 NFCORE_RNASEQ:RNASEQ:QUANTIFY_STAR_SALMON:SALMON_QUANT (R08_AM_Allo24h) COMPLETED /scratch/lianov/nfcore_rnaseq/example_run/work/e7/d392529534e631f64e6a7e6c4f0393
14/0a5cda NFCORE_RNASEQ:RNASEQ:QUANTIFY_STAR_SALMON:SALMON_QUANT (R06_AM_Allo24h) COMPLETED /scratch/lianov/nfcore_rnaseq/example_run/work/14/0a5cda0d04f9ea96035e4017029dea
7b/2d5e81 NFCORE_RNASEQ:RNASEQ:QUANTIFY_STAR_SALMON:SALMON_QUANT (R05_AM_Allo24h) COMPLETED /scratch/lianov/nfcore_rnaseq/example_run/work/7b/2d5e8145284ca8694945526209b800Choosing one specific directory from the above displays the expected files:
cd /scratch/lianov/nfcore_rnaseq/example_run/work/7b/2d5e8145284ca8694945526209b80
ls -ladrwxrwxr-x 3 lianov lianov 4096 Jul 24 19:24 ./
drwxrwxr-x 3 lianov lianov 4096 Jul 24 19:20 ../
-rw-rw-r-- 1 lianov lianov 0 Jul 24 19:21 .command.begin
-rw-rw-r-- 1 lianov lianov 8377 Jul 24 19:24 .command.err
-rw-rw-r-- 1 lianov lianov 8377 Jul 24 19:24 .command.log
-rw-rw-r-- 1 lianov lianov 0 Jul 24 19:21 .command.out
-rw-rw-r-- 1 lianov lianov 12132 Jul 24 19:20 .command.run
-rw-rw-r-- 1 lianov lianov 729 Jul 24 19:20 .command.sh
-rw-r--r-- 1 lianov lianov 279 Jul 24 19:24 .command.trace
lrwxrwxrwx 1 lianov lianov 108 Jul 24 19:21 deseq2_pca_header.txt -> /data/user/home/lianov/.nextflow/assets/nf-core/rnaseq/workflows/rnaseq/assets/multiqc/deseq2_pca_header.txt
-rw-rw-r-- 1 lianov lianov 1 Jul 24 19:24 .exitcode
lrwxrwxrwx 1 lianov lianov 117 Jul 24 19:21 gencode.vM32.annotation.filtered.gtf -> /scratch/lianov/nfcore_rnaseq/example_run/work/6e/17d80ec88c525fb159bf5433623838/gencode.vM32.annotation.filtered.gtf
lrwxrwxrwx 1 lianov lianov 102 Jul 24 19:21 genome.transcripts.fa -> /scratch/lianov/nfcore_rnaseq/example_run/work/ca/7c9423cb2c9273e9ad65c051b1bab2/genome.transcripts.fa
drwxr-xr-x 5 lianov lianov 4096 Jul 24 19:24 R05_AM_Allo24h/
lrwxrwxrwx 1 lianov lianov 127 Jul 24 19:21 R05_AM_Allo24h.Aligned.toTranscriptome.out.bam -> /scratch/lianov/nfcore_rnaseq/example_run/work/10/7419257d47c89a9a340dc08f6ff64c/R05_AM_Allo24h.Aligned.toTranscriptome.out.bam
-rw-r--r-- 1 lianov lianov 1279 Jul 24 19:24 R05_AM_Allo24h_meta_info.json
-rw-r--r-- 1 lianov lianov 77 Jul 24 19:24 versions.ymlLastly, inspecting .command.sh verifies that indeed
--seqBias --gcBias was applied to our run:
#!/usr/bin/env bash -C -e -u -o pipefail
salmon quant \
--geneMap gencode.vM32.annotation.filtered.gtf \
--threads 6 \
--libType=U \
-t genome.transcripts.fa \
-a R05_AM_Allo24h.Aligned.toTranscriptome.out.bam \
--seqBias --gcBias \
-o R05_AM_Allo24h
if [ -f R05_AM_Allo24h/aux_info/meta_info.json ]; then
cp R05_AM_Allo24h/aux_info/meta_info.json "R05_AM_Allo24h_meta_info.json"
fi
if [ -f R05_AM_Allo24h/lib_format_counts.json ]; then
cp R05_AM_Allo24h/lib_format_counts.json "R05_AM_Allo24h_lib_format_counts.json"
fi
cat <<-END_VERSIONS > versions.yml
"NFCORE_RNASEQ:RNASEQ:QUANTIFY_STAR_SALMON:SALMON_QUANT":
salmon: $(echo $(salmon --version) | sed -e "s/salmon //g")
END_VERSIONSTroubleshooting errors
Our pipeline executed successfully. While it would be ideal if every run worked flawlessly, errors are sometimes inevitable. Fortunately, Seqera AI can assist in diagnosing and resolving these issues. Below, we present two example scenarios where we intentionally introduced errors to demonstrate how Seqera AI can help troubleshoot and guide the resolution process.
Case 1
In a new run, we have encountered the following error (this can be
seen in .nextflow.log):
The following invalid input values have been detected:
* --input (./samplesheet.csv): Validation of file failed:
-> Entry 3: Missing required field(s): strandedness
-- Check script '/home/lianov/.nextflow/assets/nf-core/rnaseq/./workflows/rnaseq/../../subworkflows/local/utils_nfcore_rnaseq_pipeline/../../nf-core/utils_nfschema_plugin/main.nf' at line: 39 or see '.nextflow.log' file for more detailsAt at first glance this indicates that strandedness is
missing from our samplesheet. Let’s take a look:
sample,fastq_1,fastq_2,strandedness
N02_AM_Naive,/data/project/U_BDS/Globus_endpoints/nf-core_workshop/input/fastqs/SRX4328049_SRR7457560.fastq.gz,,auto
N01_AM_Naive,/data/project/U_BDS/Globus_endpoints/nf-core_workshop/input/fastqs/SRX4328050_SRR7457559.fastq.gz,,auto
N04_AM_Naive,/data/project/U_BDS/Globus_endpoints/nf-core_workshop/input/fastqs/SRX4328051_SRR7457558.fastq.gz,,,auto
N03_AM_Naive,/data/project/U_BDS/Globus_endpoints/nf-core_workshop/input/fastqs/SRX4328052_SRR7457557.fastq.gz,,auto
R08_AM_Allo24h,/data/project/U_BDS/Globus_endpoints/nf-core_workshop/input/fastqs/SRX4328047_SRR7457562.fastq.gz,,auto
R07_AM_Allo24h,/data/project/U_BDS/Globus_endpoints/nf-core_workshop/input/fastqs/SRX4328048_SRR7457561.fastq.gz,,auto
R06_AM_Allo24h,/data/project/U_BDS/Globus_endpoints/nf-core_workshop/input/fastqs/SRX4328057_SRR7457552.fastq.gz,,auto
R05_AM_Allo24h,/data/project/U_BDS/Globus_endpoints/nf-core_workshop/input/fastqs/SRX4328058_SRR7457551.fastq.gz,,autoAs you can see every sample does have auto indicated, so
what is the issue?
Discussion Question
Let’s spend a minutes looking over this file to see if you can spot the issue.
Now let’s see if Seqera AI can help us as well:
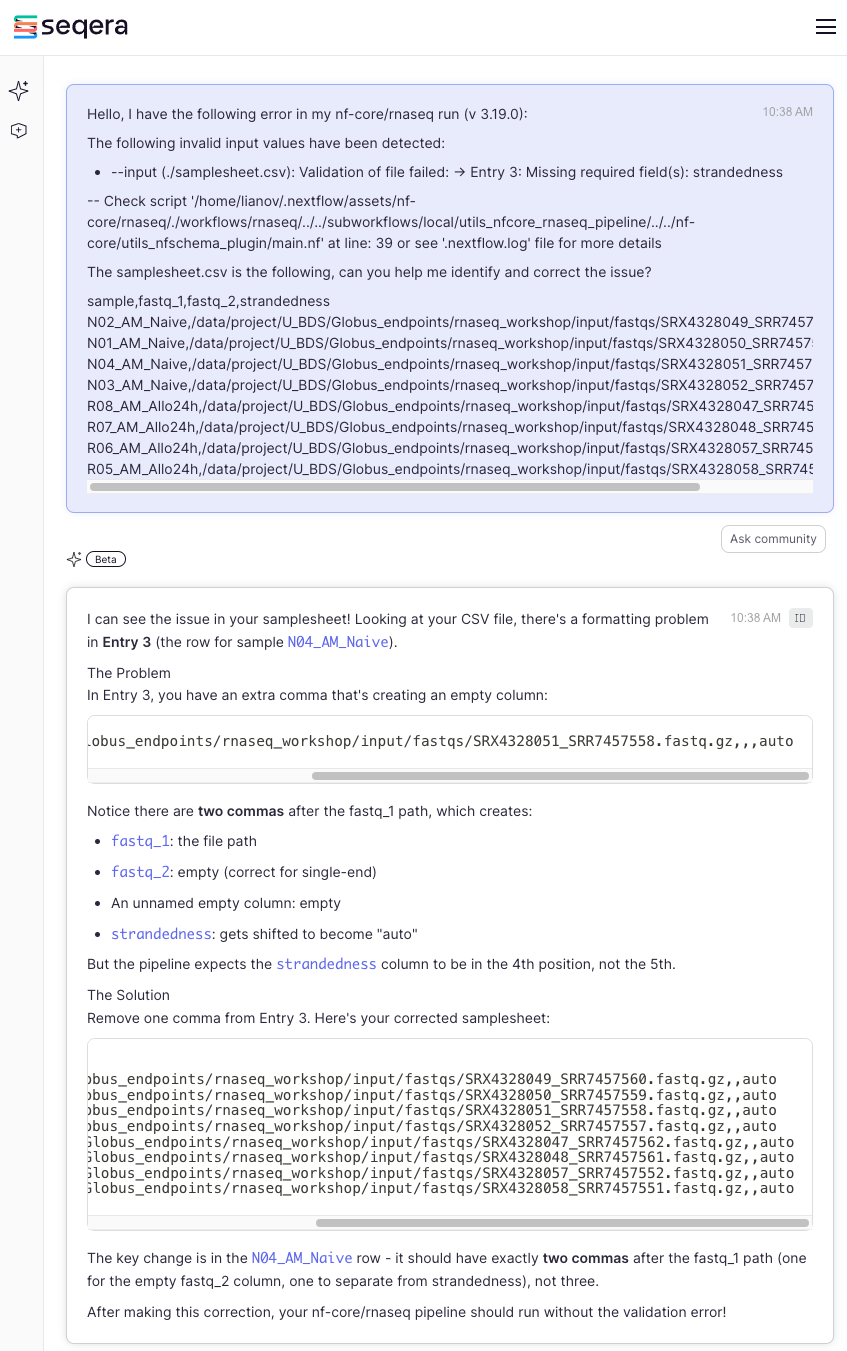
By providing the error and the samplesheet, Seqera AI was able to spot the issue right away. It provided an explanation along with the corrected samplesheet.
Case 2
Let’s try another example. In this new run, we are now receiving the following error:
-[nf-core/rnaseq] Pipeline completed with errors-
ERROR ~ Error executing process > 'NFCORE_RNASEQ:RNASEQ:QUANTIFY_PSEUDO_ALIGNMENT:SALMON_QUANT (N02_AM_Naive)'
Caused by:
Process `NFCORE_RNASEQ:RNASEQ:QUANTIFY_PSEUDO_ALIGNMENT:SALMON_QUANT (N02_AM_Naive)` terminated with an error exit status (1)
Command executed:
salmon quant \
--geneMap gencode.vM32.annotation.filtered.gtf \
--threads 6 \
--libType=U \
--index salmon \
-r N02_AM_Naive_trimmed_trimmed.fq.gz \
--seqbias --gcbias \
-o N02_AM_Naive
if [ -f N02_AM_Naive/aux_info/meta_info.json ]; then
cp N02_AM_Naive/aux_info/meta_info.json "N02_AM_Naive_meta_info.json"
fi
if [ -f N02_AM_Naive/lib_format_counts.json ]; then
cp N02_AM_Naive/lib_format_counts.json "N02_AM_Naive_lib_format_counts.json"
fi
cat <<-END_VERSIONS > versions.yml
"NFCORE_RNASEQ:RNASEQ:QUANTIFY_PSEUDO_ALIGNMENT:SALMON_QUANT":
salmon: $(echo $(salmon --version) | sed -e "s/salmon //g")
END_VERSIONS
Command exit status:
1
Command output:
(empty)
Command error:
Version Server Response: Not Found
(mapping-based mode) Exception : [unrecognised option '--seqbias'].
Please be sure you are passing correct options, and that you are running in the intended mode.
alignment-based mode is detected and enabled via the '-a' flag. Exiting.
Work dir:
/scratch/lianov/nfcore_rnaseq/example_of_error/work/e0/1bb787179e237e56d5bd2d1d38c07b
Tip: view the complete command output by changing to the process work dir and entering the command `cat .command.out`Let’s break this down first, identify the message that points to the error and then see if Seqera AI could have also helped us.
- The error message block follows a standard format for errors which
occurs within a specific process, in this case the process
NFCORE_RNASEQ:RNASEQ:QUANTIFY_PSEUDO_ALIGNMENT:SALMON_QUANT (N02_AM_Naive) - We also see that it states exit status 1. This status is a common status indicating that the job was not successful (while exit status 0 indicates success).
- It displays the command which the process executed - as we can see
this is the
salmon quantcommand which compiles the pipeline parameters along with any user-provided changes. - Command error. The
salmondevelopers created very user-friendly errors for this type of issue, where it clearly states that--seqbiasis not an option.
With --seqbias identified as the issue, what is the
solution? One point to bear in mind is case-sensitivity.
--seqbias failed because the proper parameter is
--seqBias (note the capitalized B).
Let’s see if Seqera AI could have helped us with this error as well:


As expected it solved the issue and provided additional helpful information, including:
-
Fixing the second parameter where we also introduced the same typo
(
--gcBiasinstead of--gcbias) -
Suggested the use of the flag
-resumewhich enables Nextflow to start the new pipeline run where it failed (instead of from scratch) - Suggested to check for any custom configuration files since configurations in them could be overwriting parameters in the params.yml
It did also suggest to remove these flags since they are not turned on by default. This suggestion is one where each user needs to consider it on a case by case basis. Pipeline developers typically set sensible defaults, but there are instances where enabling non-default parameters is best. Our recommendation to anyone using nf-core pipelines is to also become familiar with the underlying methods and tools - this will enable you to make the best decisions for your own data and use cases. After checking the documentation of a tool, if you have questions about specific parameters you can ask Seqera AI for help and/or the nf-core community in Slack for their input. There are also resources available online including the open-source repository for each tool (e.g.: search open/closed issues or discussions).
Considerations when using AI
While Seqera AI is a powerful tool to help you in running nf-core pipelines and programming in Nextflow, there are some important limitations to keep in mind as you use Seqera AI (and other AI tools such as ChatGPT).
Seqera AI is still susceptible to hallucinations. Hallucinations occur when an AI confidentially answers a prompt with either incorrect or entirely fabricated information. For example, we used the same prompts above and entered them into the Seqera AI extension for VSCode and received the following answers:
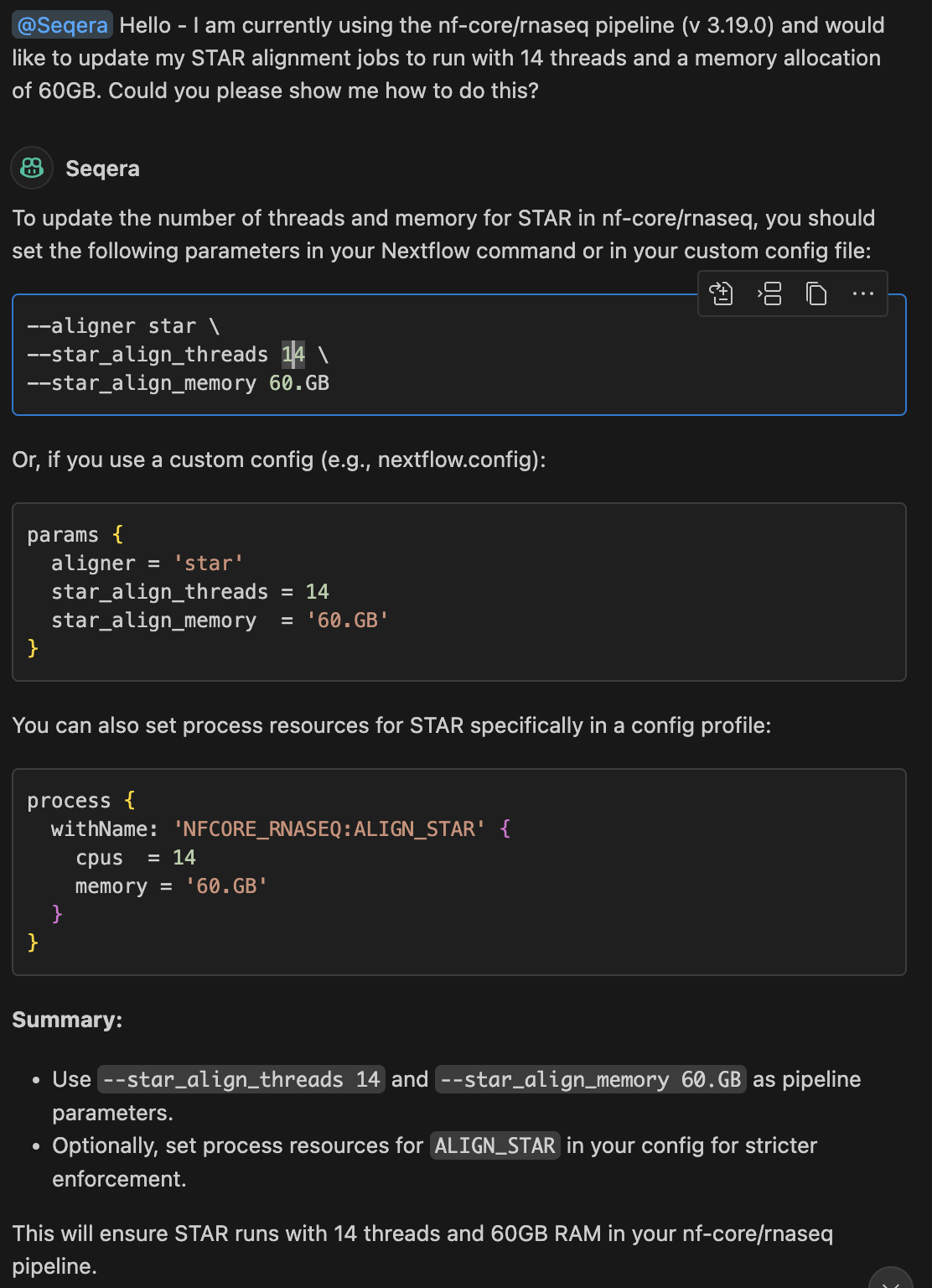
Now as you can see the Seqera AI extension generated a response that
is not entirely correct. While the last method it provided of creating a
custom configuration profile is correct, the first two methods are
entirely incorrect. The nf-core/rnaseq pipeline does not contain the
--star_align_threads or --star_align_memory
parameters, meaning these have been hallucinated.
Another consideration when using AI tools is that responses are not reproducible. AI models generate responses based on probabilistic predictions, which means that the same question asked multiple times can yield different responses. This variability can make it difficult to consistently validate or rely on a solution given by AI. To demonstrate this, we entered one of the questions above into the Seqera AI web interface again and received this response:

As you can see, even though this is the exact prompt used above, the
response is slightly different. Most notably, a description of
nextflow log is absent from the response, which is a much
easier and scalable method of obtaining information about a Nextflow
run.
Conclusion
Overall, Seqera AI is a powerful tool to use when working with Nextflow and nf-core, whether you are running a pre-built pipeline or developing your own. Because it is trained on a focused and domain-specific dataset, it often provides more relevant and accurate suggestions than generalized tools such as ChatGPT.
However, much like other AI tools, it still suffers from hallucinations and can produce inconsistent or unreproducible responses. Therefore, we recommend always double-checking any response or command generated by Seqera AI (or any other AI, for that matter). You can cross-reference responses you receive with the official nf-core documentation and community resources such as the Slack workspace or GitHub discussions.
Ultimately, Seqera AI should be seen as a helpful assistant. Use it to accelerate your work, but don’t let it be your sole source of information.